

August 2013 - New Publication from the group on The Journal of Physical Chemistry: "Ab Initio Electronic Gaps of Ge Nanodots: The Role of Self-Energy Effects"
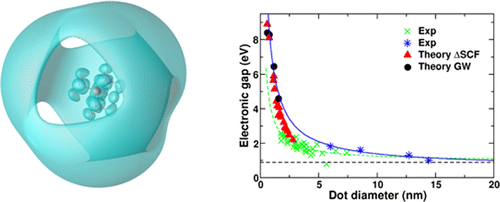
Nanostructuring of a material leads to enormous effects on its excited state properties. This study, through the application of different state-of-the-art ab initio theoretical tools, investigates the effect of size on the electronic gap of germanium nanocrystals highlighting similarities and differences with respect to equivalent silicon nanostructures. We performed both GW and ΔSCF calculations for the determination of their electronic structure. While it is known that ΔSCF corrections to the Kohn–Sham gap vanish for extended systems, the two approaches were expected to be equivalent in the limit of small clusters. However, it has been recently found that for hydrogenated Si clusters the ΔSCF gaps are systematically smaller than the GW ones, while the opposite is true for Ag clusters. In this work we find that the GW gaps are larger than the ΔSCF ones for all the Ge dots, with the exception of the smallest one. Such crossing between the ΔSCF and the GW gap values was not expected and has never been observed before. Moreover, also for hydrogenated Si nanocrystals we found a similar behavior. The origin of this crossing might be found in the Rydberg character of the LUMO of the smallest clusters and can also explain the qualitative differences in the comparison between GW and ΔSCF found in the previous studies.
- LINK: Link